 闽政通APP
闽政通APP
 微信公众号
微信公众号
美国FDA发布关于Bard公司因材料疲劳导致泄漏风险召回Power PICC中心静脉导管的警示信息
美国FDA发布关于Boston Scientific公司支架和电灼增强输送系统相关问题的早期警示信息
美国FDA发布关于Olympus公司高流量充气单元相关问题的早期警示信息
美国FDA发布关于Draeger公司因生产过程中杂质风险召回麻醉蒸发器的警示信息
澳大利亚TGA发布关于Baxter公司因组件缺陷问题召回EVO IQ大容量输注泵的警示信息
|
|
国家药品不良反应监测中心 国家药品监督管理局药品评价中心 http://www.cdr-adr.org.cn |

美国FDA发布关于Bard公司因材料疲劳导致泄漏风险召回Power PICC中心静脉导管的警示信息
发布日期:2026年1月5日
召回级别:此次召回涉及将相关器械从使用单位或经营企业移除。FDA将此次召回认定为最严重的类型。如果继续使用相关器械,可能会造成严重伤害甚至死亡。
召回器械:
4 Fr.单腔Power PICC中心静脉导管(含SOLO和非SOLO版本)
涉及器械批次列表详见FDA网站:
https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/intravascular-picc-catheter-recall-bard-removes-powerpicc-intravascular-catheters
召回原因:
Power PICC中心静脉导管适用于经外周静脉进入中心静脉系统,进行静脉输注或治疗、造影剂高压注射及中心静脉压监测,可短期或长期使用。Bard发现部分导管因材料疲劳导致的泄漏事件增加,主要表现为导管管体出现横向或环向的裂纹(如图1)。Bard调查证实,这些裂纹与制造导管时使用了存在问题的树脂材料有关。

图1 导管管体横向或环向的裂纹示例
材料疲劳导致渗漏的相关风险包括:液体外渗、组织损伤、疼痛、静脉炎、出血、空气栓塞、异物栓塞、感染及治疗中断。Bard已收到10份相关的严重伤害报告。
召回措施:
2025年3月11日,Bard向所有受影响的医疗机构发出紧急医疗器械产品召回通知,建议采取以下措施:销毁所有未使用的相关产品;如果相关产品目前正在使用中,Bard不建议移除该设备,除非怀疑发生损坏。但建议患者和医务人员应密切观察导管状态,如果发现导管断裂,应即停止输注,按照医疗机构操作指南,尽快拔除导管,并使用替代通路。
为减少使用过程中导管因材料疲劳导致泄漏事件的发生,Bard建议采取以下措施:使用带粘性背衬的固定系统代替压缩式固定系统;使用合适尺寸的固定系统,以适应锥形区域导管直径的变化;确保将导管完全插入,尽可能接近零厘米标记处(图2的B位),以使锥形区域能够发挥抗扭结的作用,以尽量降低导管渗漏率。
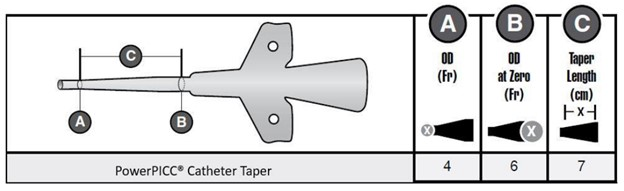
图2 Power PICC锥形区域
(美国FDA网站)
美国FDA发布关于Boston Scientific公司支架和电灼增强输送系统相关问题的早期警示信息
发布日期:2026年1月16日
这是一条医疗器械早期警示信息。FDA已经发现存在一个潜在的高风险医疗器械问题。FDA将在有重要新信息时更新此网页,使公众及时获知相关情况。
警示产品:
FDA获悉,Boston Scientific(波士顿科学)公司已经向受影响的客户发出了一封信,建议将某些支架和电灼强化输送系统从使用或销售的地方移除。
受影响的器械:
|
器械名称 |
UDI |
UPN |
|
AXIOS 支架与电灼增强输送系统 |
08714729904540 |
M00553520 |
|
AXIOS 支架与电灼增强输送系统 |
08714729904557 |
M00553530 |
|
AXIOS 支架与电灼增强输送系统 |
08714729951100 |
M00553560 |
|
AXIOS 支架与电灼增强输送系统 |
08714729951179 |
M00553660 |
|
AXIOS 支架与电灼增强输送系统 |
00191506008086 |
M00553680 |
|
AXIOS 支架与电灼增强输送系统 |
00191506008093 |
M00553690 |
详细的受影响批次请查阅FDA官网:https://www.fda.gov/medical-devices/medical-device-recalls-and-early-alerts/early-alert-stent-and-electrocautery-enhanced-delivery-system-issue-boston-scientific
器械用途:
AXIOS电灼增强支架和输送系统适用于经胃或经十二指肠内镜下对症状性胰腺假性囊肿和/或包裹性坏死的引流。某些型号也可用于高风险或不适合手术的急性胆囊炎患者的胆囊引流。
警示原因:
波士顿科学公司表示,某些AXIOS支架和电灼增强输送系统支架置入和扩展问题的不良事件出现增加趋势。这个问题发生在置入支架的过程中,并不影响已成功置入的支架。
支架放置困难可能会延长手术时间,因此可能需要更换新的支架。如果第一个凸起无法展开或扩张,则可能需要额外的内镜或手术干预来移除支架并关闭穿刺部位。
采取措施:
请勿使用或销售受影响的产品。已成功植入AXIOS支架的患者应继续遵循标准护理,因为问题只发生在置入支架时。
12月19日,波士顿科学公司向所有受影响的客户发送了一封信,建议采取以下行动:
立即停止进一步销售或使用任何剩余的受影响设备。送回波士顿科学公司前,把它们从你们工厂的库存中取出并单独放置在一个安全的地方。
立即将此信息张贴在受影响产品附近的可见位置,以确保设备的所有操作人员和用户都可以便捷地了解此信息。
与受影响产品所在组织以及已转让产品所在组织内的全部医务人员共享此通知。
如果您是经销商,则必须将此通知转发给您的客户,以确保将该通知传递给最终用户。
如果您的机构已将产品发送到其他医院或您网络内的机构,请确保将此通知转发给他们。
波士顿科学公司提醒医务人员,AXIOS支架只能按照使用说明中的指示使用。
请查看此网页了解最新情况。FDA目前正在审查有关这一潜在高风险设备的相关信息,并将在获得重要新进展时向公众通报。
截至12月23日,波士顿科学公司报告了与此问题有关的167例严重伤害和3例死亡不良事件。
(美国FDA网站)
美国FDA发布关于Olympus公司高流量充气单元相关问题的早期警示信息
这是一条医疗器械早期警示信息。FDA已经发现存在一个潜在的高风险医疗器械问题。FDA将在有重要新信息时更新此网页,使公众及时获知相关情况。
发布日期:2026年1月22日
警示产品:FDA获悉,Olympus(奥林巴斯)公司已向受影响的客户发出信函,建议将某些高流量充气单元从使用或销售地点移除。
受影响的器械:
|
器械名称 |
型号 |
序列号 |
|
高流量充气单元 |
UHI |
所有 |
|
高流量充气单元 |
UHI-2 |
所有 |
|
高流量充气单元 |
UHI-3 |
所有 |
产品用途:UHI、UHI-2和UHI-3型高流量充气单元旨在向腹腔充气,并提供自动抽吸和烟雾清除功能,以方便腹腔镜下的观察和治疗。
召回原因:奥林巴斯表示,其高流量充气单元(High Flow Insufflation Unit,型号 UHI、UHI-2 和 UHI-3)中的软件算法需要进行修正,以解决一个可能导致过压事件的潜在问题。这些产品已停产,且多年来未再提供维修支持。由于针对受影响型号无法提供纠正措施,奥林巴斯决定将这些设备退出市场。
在手术过程中,过度充气可能导致多种患者伤害,包括但不限于:空气栓塞、心律失常(如心动过缓、心搏停止或心脏骤停)、气胸、肾脏或泌尿系统问题、低氧血症、皮下气肿、治疗延误、手术复杂度增加,甚至可能导致死亡。
截至 2025 年 12 月 31 日,奥林巴斯已报告与该问题相关的2 起严重伤害事件,未报告死亡病例。
采取措施:
请勿使用受影响的设备。立即识别并隔离受影响的设备。
1月16日,奥林巴斯向所有受影响的客户发送了一封信,建议他们采取以下行动:
检查您的库存,并立即隔离任何已识别的设备。
立即停止使用您库存中的任何UHI、UHI-2和/或UHI-3。
如果您已将此产品进一步分发,请将本通知转发给其他用户和机构。
请查看FDA网页以获取最新信息。FDA目前正在审查有关此潜在高风险设备问题的信息,并将在获得重要新信息时向公众通报。
(美国FDA网站)
美国FDA发布关于Draeger公司因生产过程中杂质风险召回麻醉蒸发器的警示信息
发布日期:2026年1月16日
召回级别:此次召回包括将特定器械从使用或销售的地方移除。FDA已将此次召回确定为最严重的类型。如果您继续使用此设备而不进行纠正,可能会导致严重伤害或死亡。
召回产品:见下图

|
器械名称 |
材料号 |
UDI |
|
Draeger Vapor2000 |
M35054 |
04048675228059 |
|
Draeger Vapor3000 |
M36500 |
04048675301363 |
产品用途:Vapor 2000 和 Vapor 3000 是无需加热、经过校准的麻醉蒸发器,用于在麻醉工作站中,将干燥的新鲜医用气体与液体麻醉剂蒸汽按精确控制的浓度进行混合(富集)。
召回原因:Draeger公司表示,Vapor 2000和Vapor 3000的某一组件并未由相关供应商按照规格要求交付。在某一生产批次的蒸发器中发现了供应商生产过程中产生的杂质。
杂质(即焊接后清洁不彻底导致的金属残留物)及其与挥发性氟化气体的化学反应可能会被患者吸入或食入。风险包括肺组织损伤、气管支气管(气管和肺气道)刺激以及肺水肿。
截至12月1日,Draeger公司尚未报告与此问题相关的任何严重伤亡事件。
召回措施:
请勿使用受影响的蒸发器。
2025年11月24日,Draeger公司向所有受影响的客户发送了一封信,建议他们采取以下行动:
请勿使用受影响的蒸发器。
您当地的德尔格(Draeger)代表将会与您联系,以安排更换您的蒸发器。
如果您已将蒸发器投入临床使用,我们要求您根据使用说明书清洁麻醉设备的呼吸系统。建议使用吸气过滤器。
请确保受影响产品的所有用户以及贵组织内的其他人员均已获悉本紧急医疗器械召回通知。若您已将产品提供给第三方,请将此通知的副本转发给他们。
请至少在您的Vapor 2000/3000被更换之前,保留德尔格(Draeger)的紧急医疗器械召回通知。
(美国FDA网站)
澳大利亚TGA发布关于Baxter公司因组件缺陷问题召回EVO IQ大容量输注泵的警示信息
发布日期:2026年1月12日
召回级别:I级
召回产品:EVO IQ大容量泵
产品编号:ELVP002ANZ
序列号:SK50400101 - SK50400171
召回原因:
Baxter的投诉回顾显示,存在缺陷的门钩在输注泵生命周期的早期即发生断裂。该输注泵配有两个金属门钩,用于将泵门锁定在关闭位置。
如果金属门钩在输注开始前发生断裂,输注泵将无法启动输注,并会显示加载错误或“加载指南(Loading Guide)”界面。
如果金属门钩在输注过程中发生断裂,输注泵将发出高优先级报警【“管路进气(Air in Line)”或“泵门打开(Door Opened)”】,并中断输注。
在上述两种情况下,均需更换为一台功能正常的输注泵,才能继续或开始治疗。
截至目前,尚未收到与该问题相关的患者伤害报告。
召回措施:
只要遵循操作手册并迅速处理任何错误信息或警报,受影响的EVO IQ 大容量泵仍可继续使用。
- Baxter公司建议客户制定一个计划,以便在出现此问题时能及时更换泵。
- Baxter公司将联系每位客户,安排一次模拟临床使用的筛查,以帮助识别有缺陷的门钩。如果确认门钩有缺陷,公司将协调退回设备进行维修。
(澳大利亚TGA网站)

扫一扫在手机上查看当前页面
 闽公网安备:35010202000300号
闽公网安备:35010202000300号