 闽政通APP
闽政通APP
 微信公众号
微信公众号
内容提要
美国FDA发布关于BD公司因泄漏的风险召回SmartSite注射器套件的警示信息
加拿大Health Canada发布关于Datascope公司因电池运行时间风险召回主动脉内球囊反搏泵的警示信息
英国MHRA发布关于飞利浦公司IntelliVue MX40型号患者监护仪耗电量升高以及低电量时无报警的警示信息
美国FDA发布关于爱德华生命科学公司存在因球囊破裂的风险召回主动脉内阻断导管的警示信息
澳大利亚TGA发布关于粗毛面乳房植入物与间变性大细胞淋巴瘤的初步结果的声明
美国FDA发布关于Teleflex公司因存在断裂问题召回新生儿ConchaSmart呼吸回路的警示信息
美国FDA发布关于要求艾尔建公司主动召回BIOCELL硅胶填充毛面乳房假体和组织扩张器的公告
美国FDA发布关于BD公司因泄漏的风险召回SmartSite注射器套件的警示信息
发布日期:2019年7月1日
召回级别:美国食品药品监督管理局(FDA)将本召回识别为I类召回,是最严重的召回类型。使用这些器械可能造成严重损伤或死亡。
召回产品:BD SmartSite注射器套件
产品编号:10798696
产品编号:18046218
产品用途:BD SmartSite注射器套件与输液泵一起,以控制的方式将液体(包括药物、血液和血液产品)输送到患者体内。该泵通过输液管套件将液体注入患者的静脉或其他给药途径。
该产品主要用于为新生儿重症监护室(NICU)的患者提供关键治疗。
召回原因:BD公司正在因泄漏风险召回SmartSite注射器套件,这些设备的泄漏可能导致关键药物的注入不足,输液的延迟或中断,液体路径的污染或导致医疗服务提供者接触危险药物。
延迟输注或维持生命的药物注入不足可能导致严重的不良健康后果,特别是对微型早产儿。另外,液体通道的污染可能导致患者感染的风险增加。
到目前为止,BD公司还没有收到因该装置故障而造成严重伤亡的报告。
召回措施:BD公司向客户发送了“紧急医疗器械召回”信函,信函中指导客户:
1.立即检查其受影响产品目录和批号的库存情况,客户被要求根据其机构的程序销毁所有受此次召回影响的产品。
2.将召回信函与在其单位内使用此产品的所有医护人员分享,以确保他们知道此次召回。
3.填写所附的“客户回复表”,以便于向客户运送替代产品。
4.向FDA的Medwatch报告使用该产品对健康产生的任何不良影响。
(美国FDA网站)
加拿大Health Canada发布关于Datascope公司因电池运行时间风险召回主动脉内球囊反搏泵的警示信息
发布日期:2019年7月2日
召回产品:Datascope公司CS300、CS100、Cardiosave Hybrid IABP、Cardiosave Rescue主动脉内球囊反搏泵(IABP)
产品型号:CS300: 0998-00-3023-XX
CS100: 0998-00-0446H53 / 0998-00-0479H62
Cardiosave Hybrid和Cardiosave Rescue:
0998-00-0800-XX
召回原因:如果没有按照操作说明手册对每台IABP进行电池维护,则电池提供运行功率可能少于预期的最短运行时间。
召回措施:进行现场纠正以确保所有 IABP 用户都遵循每台设备的操作说明手册中就电池的使用、充电、维护和存储方面的建议,因为不同型号的IABP电池运行时间和放电周期不同。
(加拿大Health Canada网站)
英国MHRA发布关于飞利浦公司IntelliVue MX40型号患者监护仪耗电量升高以及低电量时无报警的警示信息
发布日期:2019年7月2日
警示原因:该设备为飞利浦生产,在使用时会比预期提前耗尽电量,但是,由于无报警,使用者可能无法觉察监护失效,这可能导致急救的延误。
采取措施:
找出所有的IntelliVue MX40设备(参见生产企业2019年6月19日更新的现场安全通报(FSNs));
如果这些设备的软件未更新至B.06.59版本,联系生产企业进行更新;
完成软件更新后:
在使用AA电池作为能量来源以及测定SpO2(手动,连续或自动模式)的监护模式下,应该每2小时更换电池;
在所有其他模式和/或使用充电电池时,应该每8小时更换电池;
按使用说明中的描述,开始使用设备前,所有的电池都应该处于充满电的状态;
根据实际情况,通过地方或国家的不良事件报告系统报告疑似或实际发生的不良事件。同时,如果地方或国家的不良事件报告系统不具备将收到的不良事件反馈给生产企业的功能,也应将该不良事件情况报告生产企业。
行动主体:所有使用和维护该设备的医生、护士以及技术人员
行动的截止日期:开始:2019年7月23日
完成:2019年8月13日。
医疗设备安全员(英格兰地区):
请生产企业将您添加至产品现场安全通报(FSNs)的发送列表。
设备详情:
涉及所有的MX40单元。可以在使用说明第15部分中找到关于原始电池预期寿命的信息。软件升级后,该性能将恢复。MX40单元见下图:
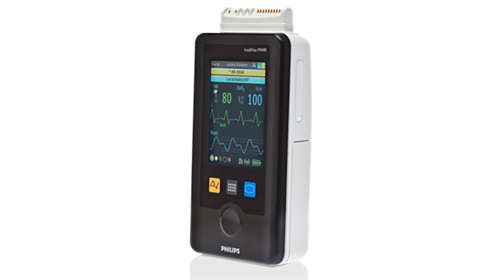
您可以通过触摸主屏幕上的电池标志,选择“设备信息”来检查每个监护仪中安装软件的版本。软件版本列在“Appl SW”下方。
生产企业联系方式:
飞利浦消费者服务中心
电话:0870 532 9741
网址:DeviceVigilanceUKI@Philips.com
(英国MHRA网站)
美国FDA发布关于爱德华生命科学公司存在因球囊破裂的风险召回主动脉内阻断导管的警示信息
发布日期:2019年7月1日
涉及器械:爱德华生命科学公司的主动脉内阻断导管(ICF100)
召回原因:由于存在球囊破裂的风险,爱德华生命科学公司主动召回了主动脉内阻断导管(型号:ICF100)UDI 编码为(01) 00690103190007,企业主动发起时间为2019年5月14日,FDA将其定义为Class I召回(最严重的级别的召回,可能导致严重伤害或死亡)。该产品一般在心肺转流术中使用,爱德华生命科学公司发现该产品在使用中存在球囊破裂的风险,导致手术时间延长,并可能导致正在施行心肺转流术的患者出现一系列的损伤,比如神经损伤、栓塞、中风,甚至死亡。在启动主动召回前,爱德华生命科学公司已经收到22个关于球囊破裂或者穿孔的个例报告,其中包括3例死亡报告。
召回措施:爱德华公司在5月14日紧急发布了召回通知,告知所有在心肺转流术中使用了该器械的患者,在5个工作日内填写相关表格反馈至爱德华公司的客服机构。同时,通知所有经销商和医疗机构收回未使用的产品。
(美国FDA网站)
澳大利亚TGA发布关于粗毛面乳房植入物与间变性大细胞淋巴瘤的初步结果的声明
发布日期:2019年7月11日
涉及器械:粗毛面乳房植入物
相关内容:由TGA宣布此次更新的声明体现了TGA在最新实验室测试和统计分析之后的初步意见。所有涉及的乳房植入物持有人可在7月24日之前提供补充资料,TGA将在达成最终决定之前评估所有企业提供的补充资料。
TGA声明的内容:(1)作为乳房植入体相关间变性大细胞淋巴瘤(BIA-ALCL)方面持续性工作的一部分,(TGA对澳大利亚市场上的毛面乳房植入体完成了审查和实验室评估。该审查于2019年5月3日公布,涉及实验室测试以及对所提供信息与乳房植入体相关间变性大细胞淋巴瘤(BIA-ALCL)已知病例的统计分析,以便估计与每种植入体类型相关的风险。(2)TGA的此次声明是基于所有已经植入粗毛面乳房植入物的患者以及考虑植入的患者都有权对其所享受的医疗保健作出知情选择,所以TGA要求所有涉及的持有人在这个阶段提供补充资料;(3)因为乳房植入体相关间变性大细胞淋巴瘤(BIA-ALCL)是罕见的,所以专家建议在植入体没有问题的情况下不要取出乳房植入体,如果有任何疑问可以咨询医生;(4)TGA的乳房植入体中心具备能力和资源,可向对其植入体有任何疑虑的患者提供指导和建议。
(澳大利亚TGA网站)
美国FDA发布关于Teleflex公司因存在断裂问题召回新生儿ConchaSmart呼吸回路的警示信息
发布日期:2019年7月17日
召回级别:美国食品药品监督管理局(FDA)将本召回识别为I类召回,是最严重的召回类型。使用这些器械可能造成严重损伤或死亡。
召回产品:具有双加热体和ConchaSmart柱的新生儿ConchaSmart呼吸回路
产品编码:870-07KIT,870-09KIT
批次信息:74L1802044,74L1802045
生产日期:2018年11月
销售日期:2018年12月至2019年1月
美国召回设备:300
启动时间:2019年5月10日
器械用途:
新生儿ConchaSmart呼吸回路适用于需要机械通气、正压呼吸辅助或一般医疗气体的新生儿和婴儿患者。该呼吸回路提供了一个在专业医疗机构内使用的病人和呼吸机之间的气体通道。该呼吸回路还包括用于哈德逊RCI海王星加热加湿器的加热电线。加热电线用于帮助维持设定的病人温度,以将呼吸机管道内的水蒸气凝结降至最低。
召回原因:
Teleflex召回具有双加热体和ConchaSmart柱的新生儿ConchaSmart呼吸回路,是因为在使用前观察到一部分旋转三通适配器存在裂纹。适配器上的裂缝可能会导致气体泄漏而无法到达患者,进而导致呼吸困难。缺氧引起的呼吸困难可能导致包括死亡在内的严重健康后果。
Teleflex收到了两份关于适配器出现裂缝的投诉。30%的适配器预计会出现裂纹。目前尚无人员伤亡报告。
可能相关的人员:
使用具有双加热体和ConchaSmart柱的新生儿ConchaSmart呼吸回路的医务人员
接受具有双加热体和ConchaSmart柱的新生儿ConchaSmart呼吸回路进行呼吸支持的患者
召回措施:
2019年5月10日,Teleflex向客户发送了一封紧急医疗设备召回信。该信指示客户:
立即停止使用并隔离库存中任何受影响的产品。
填写随函所附的确认表格,注明你是否有受影响产品的存货。请将表格传真至1-855-419-8507或发邮件至recalls@teleflex.com。
联系信息
客户如对此次召回有任何疑问或需要额外协助,请与Teleflex客户服务中心联系,电话:1-866-396-2111。
(美国FDA网站)
美国FDA发布关于要求艾尔建公司主动召回BIOCELL硅胶填充毛面乳房假体和组织扩张器的公告
发布日期:2019年7月24日
涉及器械:艾尔建公司的BIOCELL硅胶填充毛面乳房假体和组织扩张器
相关问题:基于近期收到的MDRs报告,美国FDA发现在全球范围内,由于植入艾尔建的BIOCELL粗毛面人工乳房假体,患者发生人工乳房植入物相关的间变大细胞淋巴瘤(BIA-ALCL)及其相关死亡的风险升高。该报告中,在全球范围内收集了573个BIA-ALCL病例报告,其中有33例死亡病例。在573个BIA-ALCL病例中,有481例植入的是艾尔建的BIOCELL粗毛面人工乳房假体;在33例死亡病例中,有13例植入了艾尔建的BIOCELL粗毛面人工乳房假体,(其中12例是在患者确诊BIA-ALCL时就已经获知),剩余20例尚未证实具体植入的人工乳房假体产品和品牌。上述产品均具备特殊的表面纹理,为艾尔建的BIOCELL产品独有。基于现在已经获得的信息,以及上述报告,美国FDA分析推论,艾尔建BIOCELL粗毛面人工乳房假体发生BIA-ALCL的几率大概是美国市场其他公司毛面人工乳房产品的6倍。如果继续植入该产品,可能会引起严重的、不利的健康后果,包括出现BIA-ALCL及其可能导致的死亡病例。美国FDA要求艾尔建公司主动召回在美国市场上的BIOCELL硅胶填充毛面乳房假体和组织扩张器。艾尔建公司已向FDA承诺启动上述产品的全球召回行动。
美国FDA将持续关注和继续评估其他人工乳房假体,如需要,会对其他人工乳房假体采取行动。目前认为大多数的BIA-ALCL与使用毛面乳房假体有关,特别是粗毛面人工乳房假体,例如艾尔建BIOCELL粗毛面人工乳房假体。FDA将持续评估,目前持续升高的BIA-ALCL风险是仅限于特定类型的毛面人工乳房假体,还是所有的毛面人工乳房产品均有相关性。FDA提醒所有考虑进行人工乳房假体植入的患者,要充分评估植入人工乳房假体后发生BIA-ALCL的风险。
同时FDA对已植入艾尔建BIOCELL乳房假体的患者提供以下建议:
如果没有症状,不建议移除这类或其他类型的乳房植入物,因为发展为BIA-ALCL的风险很低。如果有任何问题,请咨询您的医生;
如果遇到任何此类症状或其他变化,请向您的医生咨询是否需要进一步评估及诊断;
基于与医生的讨论结果,确认为BIA-ALCL的患者应移除植入物及周围瘢痕包膜,这是一个比单独移除植入物更大的手术。
贴士:BIA-ALCL 是一种特殊类型的非霍奇金淋巴瘤,在大多数的病例中,BIA-ALCL发生在植入物临近的疤痕组织或组织液中,也有部分出现全身侵犯导致死亡的病例报告。目前看来,BIA-ALCL总发病率较低,但是如果没有早期发现,或者患者出现迅速地全身受累,可能会导致死亡。大多数早期发现的BIA-ALCL患者可以通过局部的外科手术治疗得到有效控制,也有部分患者需要接受局部放疗或全身化疗。
(美国FDA网站)
扫一扫在手机上查看当前页面
 闽公网安备:35010202000300号
闽公网安备:35010202000300号