|

|
国家药品不良反应监测中心
国家药品监督管理局药品评价中心
http://www.cdr-adr.org.cn
|
美国FDA发布关于Asensus Surgical公司因意外移动故障问题召回 Senhance 手术系统的警示信息
发布日期:2023年11月16日
召回级别:I级
召回产品:Asensus Senhance 手术系统
召回原因:因机器人辅助手术设备发生意外移动,Asensus Surgical 公司召回Asensus Senhance手术系统。该问题表现为腹腔镜器械致动器(LIA)的手臂运动不受控制,即外科医生解除 Senhance 系统上的远程操作后,LIA 向一个方向持续旋转。Senhance 系统设计有紧急停止功能,可在发现此问题时停止操作。目前还没有病人受此影响或产生伤害,但该问题有可能造成关键组织创伤。
召回措施:2023 年 9 月 15 日,Asensus Surgical 公司向所有受影响的客户发送了一封紧急医疗器械召回信。信中要求:客户停止使用 Senhance 手术系统,直到 Asensus 服务团队将设备软件更新到 2.7.5 版本;在信中附带的 "收件确认 "文件上签名,并将其寄回 Asensus Surgical。
(美国FDA网站)
美国FDA发布关于BD公司网片产品标签更新的警示信息
发布日期:2023年11月9日
警示内容:
美国食品药品监督管理局(FDA)正在通知医疗保健提供商有关BD公司网片产品的标签更新:
Phasix网片
Phasix ST网片
Phasix插头和补丁
带开放式定位系统(OPS)的Phasix ST网状
带Echo 2定位系统的Phasix ST网状
GalaFLEX Lite支架
GalaFLEX支架
GalaFLEX 3D支架
GalaFLEX 3DR支架
这些外科用网片产品用于修复和加固存在弱点的软组织。
FDA意识到外科网片产品在乳房手术中的使用越来越多。然而,FDA尚未确定外科网片在乳房手术(包括隆胸或重建)中的安全性和有效性。
BD公司已更新这些产品的标签,包括更新的警告和预防措施。FDA发布这封信是为了确保医疗保健提供者了解这些标签更新,并了解FDA尚未确定包括BD的网片产品在内的外科网片产品在乳房手术中使用是安全有效的。
建议
FDA建议医疗保健提供者:
阅读BD的更新标签,并仔细遵循使用说明和建议。
FDA不建议无症状患者再次手术或移除植入的外科网片。
背景
FDA强调通过临床评价的手段评估这些产品用于乳房手术时的安全性、有效性和效益风险的重要性。美国FDA没有批准任何用于包括隆胸或重建在内的乳房手术的外科网片产品。
FDA行动
FDA在2019年3月25日至26日举行的普通和整形外科设备专家组会议上就此问题进行了沟通。该小组的建议没有改变。
如果有新的或额外的信息,FDA将继续通知医疗保健提供者和公众。
(美国FDA网站)
澳大利亚TGA发布关于ResMed公司因可能影响植入医疗设备功能问题召回磁吸式口罩的警示信息
发布日期:2023年11月20日
召回级别: II级
召回编号:RC-2023-RN-00941-1
召回产品:ResMed磁吸式口罩
产品批次:AirFit N10, AirFit N10 for Her, AirFit N20, AirFit N20 for Her, AirTouch N20, AirTouch N20 for Her, AirFit F20, AirFit F20 for Her, AirTouch F20, AirTouch F20 for Her, AirFit F30, AirFit F30。
注册登记号: 300769 & 300768 (Resmed Limited - CPAP/BiPAP口罩可重复使用)
召回原因:ResMed正在更新ResMed磁吸式口罩的现有禁忌症和警示信息,磁铁可能会影响某些植入医疗设备的功能,如起搏器、植入式心脏除颤器、神经刺激器、脑脊液分流器、胰岛素/输液泵和含有铁磁性材料的金属植入物/物体(动脉瘤夹/血流中断装置、栓塞线圈、支架、瓣膜、电极、用来恢复听力或平衡的植入物(植入磁铁、眼部植入物、眼内金属碎片)。
需将磁铁与植入物或可能受电磁干扰影响的医疗设备保持至少6英寸(150mm)的安全距离。
在某些情况下,当磁铁靠近某些医疗植入物/设备时,潜在的磁干扰可能会影响植入物/设备的性能或植入位置,从而可能导致严重伤害或死亡。
召回措施:ResMed正在更新客户信函中概述的现有禁忌症和警示(仅由ResMed提供给受影响的客户)。
如果患者或在使用口罩时与患者有密切身体接触的任何人(例如床伴)有客户信中的医疗植入物,他们将:
-联系口罩供应商,用不带磁铁的口罩替换带磁铁的口罩。
-请将磁吸式口罩与植入物/医疗设备保持6英寸(150毫米)的安全距离。这适用于患者和任何与磁吸式口罩有密切身体接触的人。
-如果没有其他口罩,请立即咨询医生,以确定是否可以使用其他口罩。
-在收到替代口罩后,请将磁吸式口罩处理掉。
如果客户没有ResMed磁吸式口罩的禁忌症,则可以按照客户信中更新的说明继续使用口罩。
(澳大利亚TGA网站)
美国FDA发布关于Teleflex公司因标签错误问题召回压力注射型导管套件的警示信息
发布日期:2023年11月6日
召回级别: I级
召回产品:压力注射型中心静脉导管
分销日期:2022年8月30日至2023年6月9日
美国召回数量:1905
公司发起日期:2023年8月10日
产品用途:Teleflex和Arrow International公司的压力注射型导管套件使医疗保健提供者能够接触到患者连接心脏和静脉的中央心血管系统。套件可以短期(少于30天)用于需要频繁注射或抽血的患者。有了该套件,医疗保健提供者可以进行血液采样、血压监测或输液、药物或化疗。
召回原因:Teleflex及其子公司Arrow International因以下列出的压力注射导管套件存在关于产品中氯己定存在的错误标签而进行召回。受影响的压力注射导管套件的盖子上错误地标明了产品代码和产品名称为未涂层。然而,横幅卡上正确列出了产品代码和产品名称为氯己定涂层。

被召回的设备名称和相关产品代码:
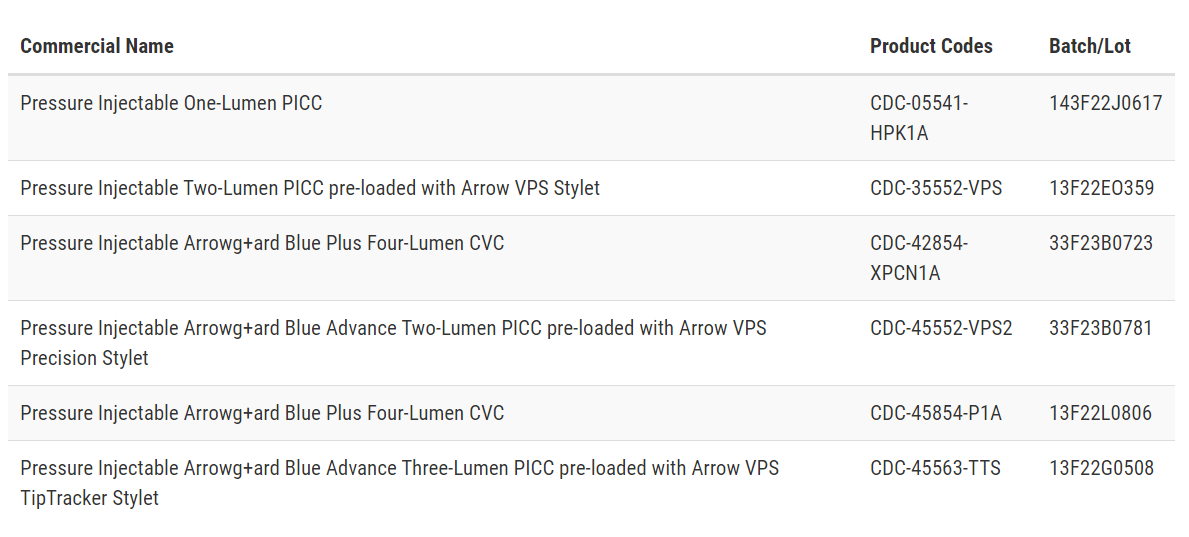
无意中使用这些套件的人可能会对健康产生严重的不良影响,如过敏反应,包括瘙痒、发红、皮肤变化、荨麻疹、头晕、血压下降、呼吸困难,以及对氯己定过敏可能导致死亡的过敏反应。
已经报告了16起与此问题相关的事件。目前还没有与此问题有关的伤亡报告。
召回措施:
2023年8月10日,Teleflex和Arrow International向医疗机构和分销商发出了一封紧急医疗器械召回信,其中包括以下建议行动:
对于医疗机构
立即:
 检查本次召回范围内的产品库存。
检查本次召回范围内的产品库存。
 停止使用和分发受影响的产品。
停止使用和分发受影响的产品。
 隔离受影响的产品。
隔离受影响的产品。
在信函随附的确认表上标记适用的复选框(受影响的产品或无受影响产品)。将表格传真至1-855-419-8507至收件人:Customer Service或通过电子邮件将表格发送至recalls@teleflex.com.
 如果您受影响的产品,客户服务代表将通过退货授权(RGA)号码与您联系,并向Teleflex提供受影响产品的退货说明。Teleflex(或当地经销商)将在收到退回的受影响产品后开具信用票据。
如果您受影响的产品,客户服务代表将通过退货授权(RGA)号码与您联系,并向Teleflex提供受影响产品的退货说明。Teleflex(或当地经销商)将在收到退回的受影响产品后开具信用票据。
对于分销商
向所有收到受影响产品的客户提供紧急医疗器械召回函的副本。每位客户必须填写确认表并将其退还给分销商。
立即停止使用和分销受影响的产品,并立即对其进行隔离。然后退回范围内的所有产品。
向Teleflex确认概述的现场活动已经完成。
完成行动后,将完成的确认表转发至recalls@teleflex.com.
如果产品在国外分销,请通知Teleflex客户服务部。
(美国FDA网站)
澳大利亚TGA发布关于Merit Medical公司因包装缺陷问题召回Merit Maestro 微导管的警示信息
发布日期:2023年11月7日
召回级别:II级
召回产品:Merit Maestro 微导管
产品编号: 28MC24150ST/F
批次编号: H2749681
召回原因: 由于供应商包装缺陷,Merit Medical Australia Pty Ltd 正在召回特定的 Merit Maestro 微导管批次。具体来说,装导管的金箔袋可能在袋子密封处有一个小孔。这个孔可能会影响设备的无菌性。
Merit 尚未收到任何与此问题相关的患者伤害/伤害报告。
召回措施:
客户应:
1. 立即确定客户信中标识的任何器械是否在其机构内。
2. 如果是这样,请隔离它们,并停止使用和分发。
3. 确保组织内的相关人员了解此现场操作。
4. 如果产品已进一步分发给其他机构或制造商,请确保立即与他们共享此通知。
5. 根据客户信函中的说明,立即将他们拥有的所有受影响的批次退还给 Merit Medical Australia。
(澳大利亚TGA网站)
美国FDA发布关于B.Braun Medical股份有限公司因报警功能故障问题召回Infusomat Space大容量输液泵的警示信息
发布日期:2023年11月17日
召回级别: I级
召回产品:Infusomat Space 大容量输液泵,无线和Infusomat Space 大容量输液泵,非无线电池组
产品型号: 8713051U and 8713052U
产品用途:Infusomat Space 大容量输液泵,包括无线和非无线Infusomat Space大容量输液泵及配套的非无线电池组,在受过专业培训的医疗专业人员在医疗机构中使用,用于成人、儿童和新生儿间歇性或连续输送静脉输液、药物、血液和血液制品。
召回原因:B.Braun Medical股份有限公司召回部分型号的Infusomat Space 容积输液泵系统,原因是其阻塞警报功能存在故障。在某些型号的产品中,可能会出现设备不存在阻塞状态时触发阻塞警报,导致输液泵停止输送药物。对于某些中断输注可能导致较高风险的药物,如血管紧张素,中断输送药物可能导致患者血流动力学不稳定,需要采取医疗措施干预以防止对患者造成身体结构或功能的永久性损害。在某些情况下,这可能是患者死因或可能导致患者死亡。B.Braun Medical股份有限公司收到与此次召回相关的投诉51起,其中包括1例严重伤害事件和1例死亡事件。
召回措施:B.Braun Medical股份有限公司向所有受影响的客户发送了一份重要医疗器械处置建议,建议所有客户:①将设备转移到只输注低风险药物的区域,不要将上述设备用于输注高风险药物。②如果正在输注高风险药物,请确保有第二个泵可替代。③该公司还将联系客户,更换合格的压力传感器。
(美国FDA网站)

 闽政通APP
闽政通APP
 微信公众号
微信公众号

 闽公网安备:35010202000300号
闽公网安备:35010202000300号
