 闽政通APP
闽政通APP
 微信公众号
微信公众号
内容提要
美国FDA发布关于美敦力公司因存在尖端损坏风险召回TurboHawk Plus定向斑块切除系统的警示信息
澳大利亚TGA发布关于GE Healthcare公司因磁铁掉落风险召回Signa & Discovery核磁共振系统的警示信息
澳大利亚TGA发布关于Getinge公司因输送系统分离风险召回Atrium Advanta V12覆膜支架系统的警示信息
美国FDA发布关于飞利浦公司因粘合剂失效风险召回Respironics V60 Plus呼吸机配件的警示信息
澳大利亚TGA发布关于B Braun公司因输注端口漏液风险召回外周/冠状动脉导管的警示信息
澳大利亚TGA发布关于Philips公司因ECG信号丢失风险召回HeartStart Intrepid监护/除颤仪的警示信息
美国FDA发布关于不要在某些美容手术中使用Renuvion/J-Plasma设备的警示信息
|
|
国家药品不良反应监测中心 国家药品监督管理局药品评价中心 http://www.cdr-adr.org.cn |

美国FDA发布关于美敦力公司因存在尖端损坏风险召回TurboHawk Plus定向斑块切除系统的警示信息
发布日期:2022年3月9日
召回级别:美国食品药品监督管理局(FDA)将本召回识别为I类召回,是最严重的召回类型。使用这些器械可能造成严重损伤或死亡。
召回产品:TurboHawk Plus定向斑块切除系统
制造日期:2021年7月21日至2021年11月25日
发行日期:2021年9月27日至2022年1月25日
在美国召回的设备:686台
产品用途:TurboHawk Plus定向斑块切除系统由导管和切割驱动器组成。该设备用于清除外周动脉堵塞和改善血液流动的手术中。
召回原因:美敦力召回(纠正)该产品是因为与另一款最近被召回进行纠正的设备具有相同的设计。在使用过程中用力时,导管内的导丝有向下移动或脱出(见下图)的风险。如果发生这种情况,导管尖端可能会断裂或分离,这可能会导致严重的不良事件,包括动脉内壁撕裂(动脉剥离)、动脉破裂、由于动脉阻塞(缺血)导致身体某个部位的血流量减少,和/或血管并发症,可能需要外科修复和附加程序来捕获和移除分离和/或移位(栓塞)的尖端。
截至2022年2月7日,没有与该问题相关的伤亡报告。
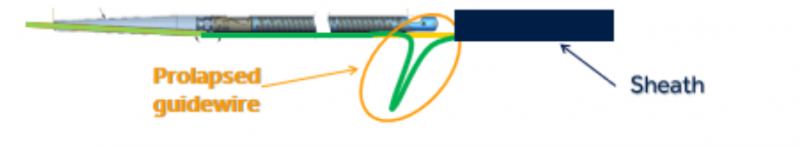
召回措施:2022年2月4日,美敦力向所有受影响的客户发送了一份紧急医疗器械通知,要求他们采取以下行动:
1.请注意,美敦力没有要求回收或处置产品。
2.与单位内部或任何存在产品转移的单位中所有需要了解的人分享此通知。
3.使用前,查看产品附带的使用说明(IFU),注意本信函中列出的警告和注意事项。
4.填写随附的客户确认表,包括紧急医疗器械通知,并通过电子邮件发送至rs.cfqfca@medtronic.com.
(美国FDA网站)
澳大利亚TGA发布关于GE Healthcare公司因磁铁掉落风险召回Signa & Discovery核磁共振系统的警示信息
发布日期:2022年3月15日
召回级别:II级
召回产品:Signa & Discovery核磁共振系统
产品用途:该产品被用于降主动脉病变的血管内修复。
召回原因:GE Healthcare提醒GE Healthcare MR系统存在潜在问题。在拆卸MR系统磁铁的过程中,如果连接到磁铁上用于运输的所有索具硬件(包括轨道和螺栓)没有正确安装和固定,可能会导致磁铁掉落,导致潜在伤害。在拆卸磁铁时,必须确保用于固定磁铁的所有硬件没有损坏,并且磁铁已被硬件正确固定。
GE Healthcare尚未收到因该问题而造成的伤亡报告。
召回措施:GE Healthcare建议客户可以继续使用其设备,并注意以下事项:
如果计划拆卸GE Healthcare MR系统,请在任何活动之前联系GE Healthcare Service(电话:1-800-659-465)或当地服务代表,以便GE Healthcare为拆卸提供指导。
GE Healthcare将向所有客户提供一份拆卸手册,其中包含有关MR系统安全拆卸的具体说明。
(澳大利亚TGA网站)
澳大利亚TGA发布关于Getinge公司因输送系统分离风险召回Atrium Advanta V12覆膜支架系统的警示信息
发布日期:2022年3月8日
召回级别:II级
召回产品:Atrium Advanta V12覆膜支架系统。
召回原因:Atrium/Getinge表示,公司在3年内收到了66起关于球囊或导管接口与输送导管分离的投诉,其中包括1起涉及肾动脉闭塞并可能导致肾功能丧失的事件。这一事件不能明确归因于球囊的分离。Atrium内部调查发现,如果在支架展开后回收输送导管时使用过大的力,可能会发生输送系统分离。
此问题是由于在移除过程中球囊中残留的液体造成的,即尝试回收时球囊未完全放气。此外,一部分Advanta V12 球囊导管尺寸可能需要比使用说明 (IFU) 中标示的更长时间来放气。
召回措施:Atrium/Getinge正在更新Advanta V12覆膜支架系统的IFU。Atrium/Getinge 建议用户可以继续使用Advanta V12覆膜支架系统,并在放气和回收设备时参考修订后的IFU信息。建议用户在给球囊放气时将充气装置上的真空拉至最大体积,并留出足够的时间进行完全放气。
(澳大利亚TGA网站)
美国FDA发布关于飞利浦公司因粘合剂失效风险召回Respironics V60 Plus呼吸机配件的警示信息
发布日期:2022年2月25日
召回级别:美国食品药品监督管理局(FDA)将本召回识别为I类召回,是最严重的召回类型。使用这些器械可能造成严重损伤或死亡。
召回产品:飞利浦Respironics V60 Plus呼吸机配件
产品编号:1138747
召回原因:Respatrics California有限责任公司发现部分呼吸机是用过期的粘合剂组装的。如果粘合剂失效,支架可能会松动,并可能损坏电容器,从而导致呼吸机停止为患者提供通风。该故障可能会激活视觉和听觉报警,也可能不会发出或显示报警(无声关机)。
召回措施:2022年1月24日,该公司通过优先邮件向客户发送了一封“紧急医疗器械纠正”信函,通知他们,已确定部分呼吸机内部部件装配时使用过期粘合剂。有可能同时发生两种故障,这两种故障由粘合剂故障激活,并且如果组装粘合剂的部件也发生故障。故障的结果可能会导致:①呼吸机停止运行,激活视觉和听觉警报,或②呼吸机停止运行,不激活视觉或听觉警报,导致“无声关机”。Respatrics California指导客户:①将其设备的序列号与附件A:受影响序列号列表进行比较,以确定呼吸机是否受到影响。设备序列号信息可位于呼吸机后部。或者,当呼吸机运行时,可以从显示屏上查看呼吸机的序列号。选择屏幕底部的菜单选项卡,然后选择通风信息。②客户应采取的其他措施:无需停止使用受影响的呼吸机,呼吸机具有远程报警系统功能,可将呼吸机连接至远程报警系统,公司建议使用远程报警器。即使呼吸机的主报警系统没有报警,远程报警也会提供备用报警。连接远程报警系统的说明见操作手册B-5节:“远程报警端口”,务必遵循操作手册和紧急医疗器械纠正函中的说明,以进一步降低与此潜在故障相关的任何风险。操作手册的提示:①提供外部氧气监测,在氧气供应损失或呼吸机故障的情况下将患者风险降至最低;②及时应对所有低风险。
(美国FDA网站)
澳大利亚TGA发布关于B Braun公司因输注端口漏液风险召回外周/冠状动脉导管的警示信息
发布日期:2022年3月11日
召回级别:II级
召回编号:RC-2022-RN-00455-1
召回产品:外周/冠状动脉导管,输注
产品批号:多个产品和批号
产品注册号:ARTG 135323 (B Braun Australia Pty Ltd -外周血管/冠状动脉导管,输注)
召回原因:B Braun公司发现一些外周/冠状动脉导管的注射端口可能会漏液。
该缺陷可能会对患者造成潜在的严重的临床后果,例如失血、药物剂量不足或延迟治疗等。用户或患者会因接触不相容物质或外来血液而处于危险之中。
召回措施:
建议客户立停止使用相关产品。
可对相关物品进行检验和退货,也可在现场销毁,只要提供销毁证明即可。
(澳大利亚TGA网站)
澳大利亚TGA发布关于Philips公司因ECG信号丢失风险召回HeartStart Intrepid监护/除颤仪的警示信息
发布日期:2022年3月3日
召回级别:I级
召回产品:HeartStart Intrepid监护/除颤仪,产品型号867172,制造日期2019年12月2日至2021年11月24日。
召回原因:
Philips意识到HeartStart Intrepid的12导联ECG功能在(a)连接到患者和(b)右腿连接变得断断续续时,可能会丢失ECG信号。这种ECG信号丢失发生时可能不会产生“leads off”警告,设备所有通道上的ECG显示为虚线。
此问题出现在(a)最初将导联电极放置在患者身上以捕获12导联ECG和(b)在12导联ECG监测患者时间歇性发生。如果在放置ECG导联时发生错误,监护仪显示屏将显示“leads off”或显示屏的所有通道上会出现一条虚线。如果在12导联监测时间歇性发生此问题且没有生成“leads off”警告,那么除非用户正在积极观察屏幕上的12导联心电图,否则可能无法发现。
迄今为止,已经报告了一起可能与此问题有关的死亡事件。
召回措施:
Philips正在开发针对此问题的解决方案,并将在2022年第二季度发布后立即安排永久性解决方案。
作为过渡,Philips建议客户按照IFU中的说明继续使用受影响的设备,并采取客户信中描述的额外预防措施(客户信将仅发送给受影响的客户)。
Philips还建议客户如果患者使用其他设备获取12导联心电图,则应确保intrepid不连接到交流电源,因为外部监视器可能会因此出现干扰。相反,请确保intrepid使用电池供电。
(澳大利亚TGA网站)
美国FDA发布关于不要在某些美容手术中使用Renuvion/J-Plasma设备的警示信息
发布日期:2022年3月14日
警示产品:Apyx 医疗的Renuvion/J-Plasma系统
产品用途:Apyx 医疗的Renuvion/J-Plasma系统包括等离子/射频机头和等离子发生器,是经批准用于普通外科手术的医疗设备。这些设备使用射频(RF)能量和氦气来产生等离子体(具有高热的气体状物质)。在手术过程中,等离子体可用于切割、凝固(止血)和加热消除软组织。该装置在任何特定手术(包括皮肤美容手术)中使用的安全性或有效性尚未确定。
警示内容:美国食品药品监督管理局(FDA)警告消费者和医疗服务提供者不要将Apyx 医疗公司的Renuvion/J-Plasma设备用于某些美容手术。具体而言,FDA警告不要用于通过皮肤表面重建(一种在皮肤上治疗皱纹的方法)或皮肤收缩(一种在皮肤下进行的方法,可以单独进行或与吸脂术结合进行以实现皮肤效果,如“收紧”)来改善皮肤外观的方法。
Renuvion/J-Plasma设备经美国食品和药物管理局批准,可在开放手术和腹腔镜手术中用于软组织的切割、凝固和消融。对于任何旨在改善皮肤外观的特定程序,尚未确定使用该装置是否安全或有效。FDA已经收到Renuvion/J-plasma设备直接用于皮肤时的严重不良事件,以及在皮下使用时的具有潜在生命威胁的不良事件。
给消费者的建议
l 请注意,Renuvion/J-Plasma设备用于任何改善皮肤外观的美容手术尚未获得FDA的批准或认可。
l 与您的医疗服务提供者讨论所有可用的皮肤美容手术的益处和风险。
l 如果您正在考虑任何皮肤美容手术,请询问您的提供商是否计划在手术过程中使用Renuvion/J-Plasma设备。
l 如果您正在考虑吸脂,请询问您的医疗服务提供者是否计划在手术过程中使用Renuvion/J-plasma设备。
l 如果您在使用Renuvion/J-Plasma后遇到任何问题或担心,请向有执照的医疗服务提供者寻求帮助。
l 向FDA报告使用Renuvion/J-Plasma程序中遇到的任何问题或并发症。您的报告与其他来源的信息,可以提供用于改善患者安全的资讯。
对医疗保健提供者的建议
请注意,FDA未批准Renuvion/J-Plasma使用于任何皮肤美容手术。
请注意,使用Renuvion/J-Plasma进行皮肤美容手术可能会导致严重且潜在危及生命的不良事件。
请勿单独使用Renuvion/J-Plasma设备或将其与吸脂术结合使用进行皮肤表面修复或皮肤收缩。
与你的病人讨论所有可用的皮肤美容手术的受益和风险。如果您正在进行美容手术,请告知您的患者您计划使用哪些设备。
查看Apyx Renuvion/J-血浆标签和用户手册,以正确使用本医疗器械。
向FDA报告使用Renuvion/J-Plasma过程中患者遇到的任何问题或并发症。
美容皮肤手术中使用Renuvion/J-Plasma的相关风险
FDA已经收到报告,描述了该装置用于皮肤美容手术后出现的严重且可能危及生命的不良事件。报告的事件包括二度和三度烧伤、感染、皮肤颜色改变、疤痕、神经损伤、大出血以及皮肤下、体腔和血管中的空气或气体积聚。在某些情况下,不良事件需要在重症监护室(ICU)治疗。
FDA行动
FDA正与制造商合作,评估所有关于Renuvion/J-Plasma用于皮肤美容手术的可用信息,并通知患者和医疗服务提供者,该设备尚未被确定为对这些手术安全或有效。
FDA将继续监测不良事件的报告。如果有新的重要信息,FDA将及时通知公众。
(美国FDA网站)

扫一扫在手机上查看当前页面
 闽公网安备:35010202000300号
闽公网安备:35010202000300号